Hello, my dear friends!
@dexterdev is back after a long gap.
Today I will discuss on creating infinite molecular structures for MD simulations!!! (Sounds impossible task... Right?)
In MD(molecular dynamics) forums etc, you will find these queries very often. For example:
- See this research gate link: How to create an infinite graphene sheet with proper periodicity?.
- Another link from namd-l forums: Infinite Armchair Single Wall Carbon Nanotubes and Periodic Boundary Conditions (PBC)
So I thought why not document this problem with a solution on STEEM blockchain.
There are times when you require to create infinite molecular structures("infinite" in the sense that the molecules in one simulation cell are covalently bonded at the interface to the respective molecules in the adjacent periodic images) in MD simulations. For example, if you want to learn how a DNA chain or protein interacts with a Carbon nanotube, you may want to keep this particular nanotube structure span whole periodic cells. It seems impossible at first sight. But there are some hacks through which we can create such structures. I will explain this in the context of VMD and NAMD softwares. As I have explained in my previous articles, VMD is a visualization software for molecular dynamics(MD) trajectories and NAMD is a MD engine.

Different periodic images of an all-atom simulation system, where the carbon nanotube(cyan) technically spans infinitely(in the sense the nanotube is covalently bonded at the interface of different periodic images) in the z-direction(blue axis). The white transparent box is waterbox and the purple molecule is a protein.
Repository
https://github.com/dexterdev/STEEMIT/tree/master/INFINITE_CNT
What will I learn?
You will learn how to set up a molecular dynamics system where molecular structures should extent in its periodic cells infinitely.
Softwares used: VMD, NAMD
Necessary Plugin for connecting bonds between periodic images: topotools
(Topotools comes with latest versions of VMD)
If you are not familiar with VMD and NAMD etc, please follow my past articles. Links in References.
Difficulty
Intermediate
System
The System comprises of:
- Carbon nanotube(armchair CNT(5,10))
(5,10) are chirality numbers. It is of no significance here for this article. - Aβ intrinsically disordered protein(from 1Z0Q.pdb)
- Waterbox with TIP3P water model
- NaCl ions with concentration 150mM/litre to neutralize the total charge in system
Tutorial Content
This tutorial is broken down into the following sections:
- Creating CNT psf,pdb files using Nanobuilder VMD plugin
- Adding the IDP(intrinsically disordered protein) to the system and positioning it properly
- Adding Waterbox and ions
- Using topotools to modify the structure by adding bonds in CNT C atoms between self and periodic image
- Equilibration RUN setup
- Verifying Carbon bonds between self and periodic cell are functioning properly
Creating CNT psf,pdb files
This is pretty straightforward. Open Carbon Nanostructure builder tool from VMD>Extensions>Modeling.
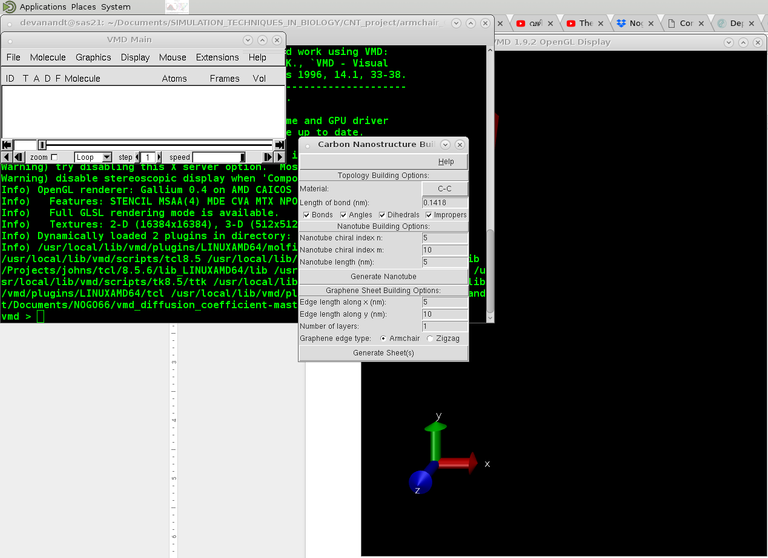
Choose CNT options accordingly. Chirality numbers and length of tube etc. Once the structure is created, create the pdb psf files using:
animate write pdb CNT.pdb
animate write psf CNT.psf
in VMD Tk console.
Adding the IDP to the system and positioning it properly
The IDP structure can be downloaded from PDB databank(LINK). The PDB id is 1Z0Q. Load the CNT structure and you can position the loaded IDP wherever appropriate(VMD hotkey for moving molecule is 8). Merge the structures in VMD. Save the merged structures.
Adding Waterbox and ions
package require solvate
solvate ./OUT.psf ./OUT.pdb -minmax {{-30.56800079345703 -11.4019999504089355 0.0} {40.322999954223633 47.20600128173828 119.11799621582031}} -o OUT_water -s WB
The above script can be used to add water. Change the coordinates appropriately. Add ions using Autoionize from here VMD>Extensions>Modeling.
Using topotools
There is a major issue in the structure we have created now. The CNT terminates at box edges of the "self" simulation cell. Which means there is a minor gap between this structure and the CNT in the next periodic cells. Topotools can be used to modify the structure by adding bonds in CNT C atoms between self and periodic images.
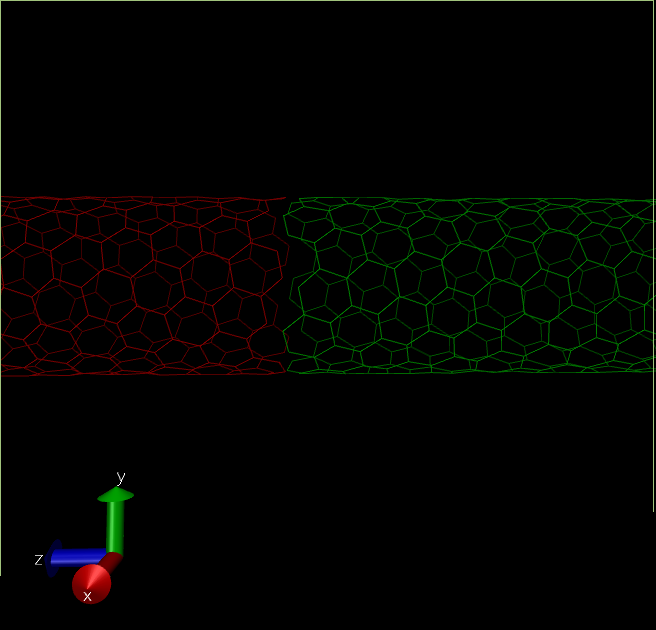
(self-red,periodic-green. See there is no bond between red and green tubes, which can create unnecessary artifacts.)
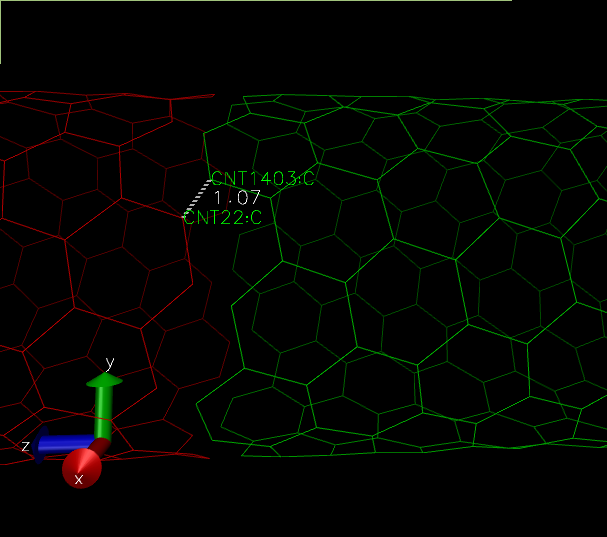
(The distance between the "supposed to be" carbon-carbon bond, is lesser than the usual 1.4 Angstroms.)
So how do we create infinite CNT is the question. There is a hack possible with the aid of topotools. Topotools can create bonds between atoms. The format is as below:
package require topotools
topo addbond $atom-index1 $atom-index2
For example in the above figure, we need a bond between atom 22 and 1403. The respective atom indices are 21 and 1402.
topo addbond 21 1402
You do this for the multiple selections. To make things easier in terms of getting at least the required atom indices, you can use the below selection commands(The selections lie in the 0.9 Angstrom edges of the nanotube):
set sel [atomselect top “segname TUB and z<0.9”]
$sel get index
set sel [atomselect top “segname TUB and z>122.5”]
#(118.2 = 123.2-0.9)
$sel get index
After applying that topo command, let us see the image:
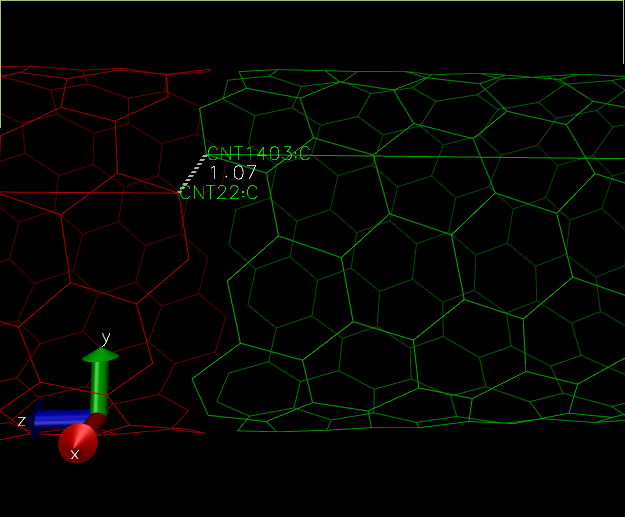
It seems like something has gone wrong. The bond went in between self molecules rather than between self and periodic cell. This problem which is a VMD rendition one will persist in our simulation. But we can verify if there is a proper bond between self and periodic cell structure by running a small simulation. We will come to that section later.
Equilibration RUN setup
structure ./OUT_water_ionized_bondcorrection.psf
coordinates ./OUT_water_ionized_bondcorrection.pdb
set temperature 298
set outputname test
firsttimestep 0
#############################################################
## SIMULATION PARAMETERS ##
#############################################################
# Input
paraTypeCharmm on
parameters ./par_water_ions.prm
parameters ./par_nanotubes.inp
parameters ./par_all36_prot.prm
temperature $temperature
# Force-Field Parameters
exclude scaled1-4
1-4scaling 1.0
cutoff 12.
switching on
switchdist 10.
pairlistdist 15.0
margin 8.0
# Integrator Parameters
# fullElectFrequency*timestep <=4.0
# stable time steps:
# with rigidBonds=all: electro:6fs;short-rangeNB:2fs;bonded:2fs
# otherwise : 2fs 2fs 1fs
timestep 2.0 ;# 2fs/step
rigidBonds all ;# Needed for 2fs steps
useSettle on
nonbondedFreq 1
fullElectFrequency 2
stepspercycle 10
zeroMomentum yes
# Constant Temperature Control
langevin off ;# do langevin dynamics
langevinDamping 5 ;# damping coefficient (gamma) of 5/ps
langevinTemp $temperature
langevinHydrogen off ;# don't couple langevin bath to hydrogens
#temperature coupling for temperature coupling
tCouple on
tCoupleTemp $temperature
# Periodic Boundary Conditions
# center of the periodic box; NOT ORIGIN!!!
cellBasisVector1 71 0 0
cellBasisVector2 0 68 0
cellBasisVector3 0 0 124
cellOrigin 10.136194229125977 18.867944717407227 61.83677673339844
#add appropriate numbers at # above
wrapAll on
# PME (for full-system periodic electrostatics)
# multiples of 2,3,5 & >=dimensions above
PME yes
PMEGridSizeX 90
PMEGridSizeY 90
PMEGridSizeZ 150
#add appropriate numbers at # above
# Constant Pressure Control (variable volume)
useGroupPressure yes ;# needed for rigidBonds
useFlexibleCell no
useConstantArea no
langevinPiston on
langevinPistonTarget 1.01325 ;# in bar -> 1 atm
langevinPistonPeriod 100.
langevinPistonDecay 50.
langevinPistonTemp $temperature
# Output
outputName $outputname
restartfreq 50000
dcdfreq 10000
xstFreq 10000
outputEnergies 10000
outputPressure 10000
#############################################################
## EXECUTION SCRIPT ##
#############################################################
# Minimization
minimize 5000
reinitvels $temperature
run 111000 ;# 222ps
Verifying Carbon bonds between self and periodic cell are functioning properly
To verify the topotools hack, we can check the the evolution of carbon-crbon bonds in the CNT at the interface. Between our indices 21 and 1402, the bond distance starts at 1.07 Angstroms. It spikes to 1.57 at minimization and evolves towards 1.44 which is great! Now once you feel the system is equilibrated, you can restart the simulation with fixing the CNT atoms in simulation setup.
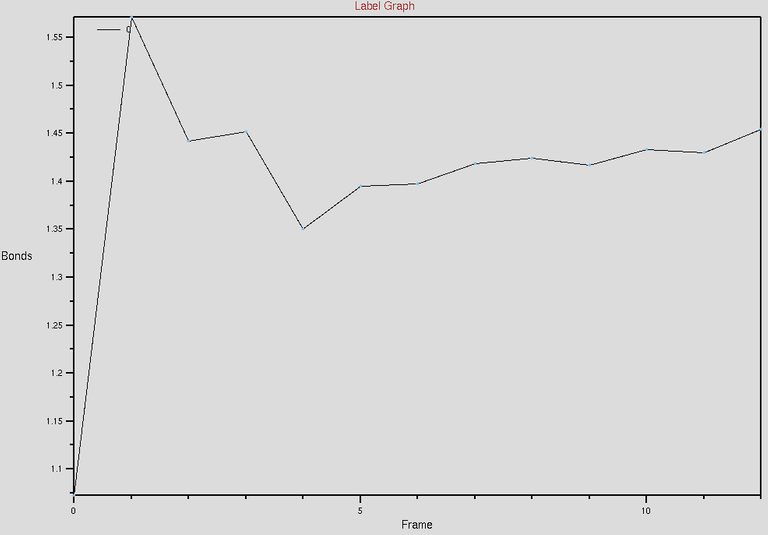
The above figure shows the evolution of C-C (between 21 and 1402 indices) bond distances w.r.t time. The time we simulated for is around 222ps. See the distance between carbon-carbon bonds saturating towards 1.44-ish Angstroms, which is what we need.

(Evolution of distances - A gif.)
Necessary Files
All necessary files are uploaded in Github.
LINK: https://github.com/dexterdev/STEEMIT/tree/master/INFINITE_CNT
References
My previous posts:
To learn about VMD and PDB file format, see here:
- https://steemit.com/steemstem/@dexterdev/visualizing-bio-molecules-in-computer-part-1-let-us-inspect-a-pdb-file-and-see-it-using-vmd
- https://steemit.com/steemstem/@dexterdev/visualizing-bio-molecules-in-computer-part-2-introduction-to-tcl-scripting-environment-in-vmd-1-sbd-prize-task-inside
To learn about the concepts in All-atom molecular dynamics see articles below:
- https://steemit.com/steemstem/@dexterdev/classical-molecular-dynamics-series-part-1-the-fundamentals
- https://steemit.com/steemstem/@dexterdev/classical-molecular-dynamics-series-part-2-the-force-field
- https://steemit.com/steemstem/@dexterdev/classical-molecular-dynamics-series-part-3-solving-the-molecular-dynamics-equation
To setup and run simulations in NAMD software, see below:
- https://steemit.com/steemstem/@dexterdev/classical-molecular-dynamics-series-part-4a-let-us-setup-a-simulation-and-run-it
- https://steemit.com/steemstem/@dexterdev/classical-molecular-dynamics-series-part-4b-running-small-systems-on-your-computer
- https://steemit.com/steemstem/@dexterdev/let-us-cool-dmpc-bilayer-lipids-an-18-day-long-molecular-dynamics-experiment-on-hpc-facility
Textbook references for learning theory of Molecular Dynamics:
- "Statistical Mechanics: Theory and Molecular Simulations" by Mark E. Tuckerman
- "Molecular Modelling: Principles and Applications" by Andrew R. Leach
- "Computer Simulation of Liquids" by D. J. Tildesley and M.P. Allen
References specific to NAMD and VMD:
#steemSTEM
#steemSTEM is a very vibrant community on top of STEEM blockchain for Science, Technology, Engineering and Mathematics (STEM). If you wish to support steemstem visit the links below:

Quick link for voting for the SteemSTEM Witness(@stem.witness)
Delegation links for @steemstem gives ROI of 65% of curation rewards
(quick delegation links: 50SP | 100SP | 500SP | 1000SP | 5000SP | 10000SP).
Also visit the steemstem app here: https://www.steemstem.io
Follow me @dexterdev
____ _______ ______ _________ ____ ______
/ _ / __\ \//__ __/ __/ __/ _ / __/ \ |\
| | \| \ \ / / \ | \ | \/| | \| \ | | //
| |_/| /_ / \ | | | /_| | |_/| /_| \//
\____\____/__/\\ \_/ \____\_/\_\____\____\__/
 credit: @mathowl
credit: @mathowl
I can't claim I know much what this is about... I guess the title says it all...
It's probably self-explanatory, just way above my head... but it looks effin intriguing! Where are those #steemSTEM curators at?!
Thank you for the encouragement @fraenk . I highly appreciate it. Thanks for visiting. But I guess this post may not attract that many popsci readers. It is not accessible to most people. Perhaps that's why it got no recognition. :P
Update: I got steemSTEM vote finally😍
Nice article.
But your coordinate axes are confusing me.
X axis is the frames which can be converted to time.
Y axis is in Angstrom units. (since they are in bond distance units)
Let me know if you have specific doubts. I will clarify it.
Hm. I simply associated it with a Cartesian coordinate system. Which is probably why nobody else questioned the coordinates.
I found myself trying to sort out where z should point to if x was my right thumb, y and z being the second and third finger, respectively.
Well, you probably should ignore my thoughts, I'm afraid I'm being ridiculous. I just tried to figure out the orientation with my left hand too.
This post has been voted on by the SteemSTEM curation team and voting trail in collaboration with @utopian-io and @curie.
If you appreciate the work we are doing then consider voting all three projects for witness by selecting stem.witness, utopian-io and curie!
For additional information please join us on the SteemSTEM discord and to get to know the rest of the community!
Hi @dexterdev!
Your post was upvoted by Utopian.io in cooperation with @steemstem - supporting knowledge, innovation and technological advancement on the Steem Blockchain.
Contribute to Open Source with utopian.io
Learn how to contribute on our website and join the new open source economy.
Want to chat? Join the Utopian Community on Discord https://discord.gg/h52nFrV
Congratulations @dexterdev! You have completed the following achievement on the Steem blockchain and have been rewarded with new badge(s) :
Click here to view your Board of Honor
If you no longer want to receive notifications, reply to this comment with the word
STOPDo not miss the last post from @steemitboard:
Congratulations @dexterdev! You have completed the following achievement on the Steem blockchain and have been rewarded with new badge(s) :
Click here to view your Board of Honor
If you no longer want to receive notifications, reply to this comment with the word
STOPDo not miss the last post from @steemitboard:
Congratulations @dexterdev! You have completed the following achievement on the Steem blockchain and have been rewarded with new badge(s) :
Click here to view your Board of Honor
If you no longer want to receive notifications, reply to this comment with the word
STOPDo not miss the last post from @steemitboard: