The following is a report I wrote for instrumental analysis in the spring of 2016. At least one of these was due every week, and sometimes the number was as high as three a week.
Now we get to see why so many chemistry students drop out; this is B minus work.
This is my old work, I am the original author; I don't think it'll set off a plagiarism checker but just in case I wanted to make sure that's known. I've left any sort of grammatical error intact, bc realistically these things will miss your radar when you're putting out this kind of volume in school
Dead Name
Electronic Transitions of Common Organic Compounds
2/3/2016
Due 2/10/2016
I. Introduction
Purpose
To quantitatively characterize and investigate compounds, some form of spectroscopy is almost inevitably required. Though each spectroscopic technique varies depending on the wavelength of light it tests for, the general principle remains the same; a source material emits photons of a specific energy level that, if they precisely match the difference between one electron level and another in a molecule, will be absorbed by said electron1,2. What occurs afterwards is dependent on both the sample in question and the type of spectroscopy; reported here is a procedure for utilizing UV-Vis range spectroscopy to determine the extinction coefficient of various solutions, along with a discussion of the theory behind it along with its limitations.
UV-Vis Spectroscopy: Overview
The UV-Vis region is defined as the region of light with a wavelength of 400 to 10 nanometers. In UV-Vis spectroscopy, a blank is taken by shining a beam of light through a cuvette containing only the solvent to be used in the sample solution; the percent absorbance of the wavelengths of light absorbed by this solvent are subtracted from the sample readings2. Following this, a beam of light is shone through a sample; during this process specific photons of light will be absorbed by the method lain out in the preceding paragraph. The same beam of light is measured on the opposite end of the sample, and after correcting for the absorbance of the solvent, the wavelengths that are “missing” have been absorbed by the sample. It might seem intuitive that this is the only information needed to determine the nature/identity/concentration of the sample, and while it is certainly a component of the work necessary, there are several other factors to consider.
Beers Law
Beer’s Law is a postulate that states that absorption of light at a given wavelength by a sample is linearly related to its concentration times the pathlength (length of the container)1,2. Mathematically, this is represented as Absorbance = L c, where is the molar extinction coefficient and L is the path length of the cell holder. is a constant unique to each solution (including different concentrations of the same solution) and is the ratio of the absorbance to the pathlength times the concentration; in other words, the amount absorbed per unit concentration. . From this information, it is intuitively obvious that the absorbance of a solution at lambda max is directly related to the concentration of the solution, and indeed from the information provided by Beers Law we can (under ideal conditions) plot a linear graph of absorption vs. concentration. Deviations do exist however. The lower the concentration, the higher the extinction coefficient; ideally the relation between the two should be linear as well, but sample interaction as well as solvent effects can cause a negative deviation from the expected value of
The Solvent Effect & Concentration Effect
The chemistry of spectroscopy and the reason for the observed absorption will be elaborated on in further sections, however it is first necessary to explore what is known as the solvent effect. Any pure compound will have characteristic absorption peaks that are repeatable and unchanging. Upon making a solution of them, these shifts can be altered depending on the nature of the solvent; i.e., a compound that is highly polar and relatively small can form solvation shells with a solvent such as water, which stabilizes the outer electrons in question and lowers the energy gap between its outer orbitals, thus producing peaks at a higher wavelength (redshift)3. Likewise, n- π* transitions can experience an increase in energy and a decrease in wavelength (blueshift) by interacting with polar solvents; this phenomenon is due to the solvent aligning itself with the ground state and thus lowering its energy which raises the gap between n and π*3. By no means is this a conclusive list of solvent effects, and relevant examples will be highlighted as they occur. Deviations from Beers law also occur at high concentrations (c>.01M)4 due to sample-sample interaction such as dimerization or electrostatic interactions, as these factors can lower the energy gap between orbitals and thus cause a negative deviation from Beer’s law.
The Chemistry of UV-Vis
For UV-Vis spectroscopy we are concerned with 4 types of electron transfer: σ- σ* (require large amounts of energy and are consequently observed in the far UV region below 150 nm), n- σ* (150-250 nm range and occurs in saturated compounds with unshared electrons), n- π* and π- π, occur in the 200-700 nm range and indicate the presence of at least one double bond, with n- π also requiring unshared electrons. These numbers are not random; the energy difference between the sigma and sigma antibonding orbitals is the greatest of the 4 sets and as such requires the most energy. The same logic can be used to make sense of the remaining three, and the following Jablonski diagram5 provides an illustration of these energy gaps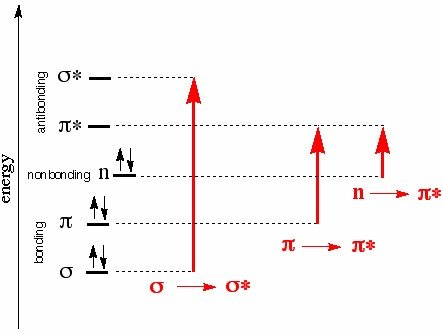
Figure 1, Jablonski diagram showing energy vs transition type
Two questions remain: What part of the molecule is actually absorbing this energy and can this be affected by intramolecular causes? Bonding electrons in a molecule, conjugated portions of a molecule, and localized nonbonding outer electrons can all absorb light strongly in the UV region, and the area which causes this absorption is known as a chromophore. Portions of the molecule which don’t absorb strongly in the UV region yet still influence the wavelength of absorption are known as auxochromes. An example is of this influence occurs in diphenyl, which due to being highly conjugated and thus having lower energy gaps between its bonding and nonbonding orbitals, has a relatively high wavelength of peak absorption. Note, however, that the addition of methyl groups in both ortho positions interferes with the planarity of the molecule due to steric hindrance which consequently interferes with the conjugation effect, causing a blueshift. Auxochromes cause redshifts as well; polar functional groups such as OH, O-, and so on can interact with and stabilize the electron cloud of the chromophore in π- π* transitions.
II. Instrumentation
Diagrams and Instrumentation
For double beam UV-spectrometers, the general components and their arrangement are as shown in figure 3, and the layout of the machine used in this procedure is shown in the drawing above it. Light is first produced by a suitable source. The light then disperses in or on the monochromater (composed of subunits elaborated on below) and, in the case of the double beam UV-spectrometer, is sent through a beam splitter (in our case a rotating chopper is used). From here, the light reflects off of a series of mirrors until it passes orthogonally through the cuvettes which contain your sample and “blank”. Specific wavelengths are absorbed per the mechanisms outlined in the previous section, and the remaining light passes through a photo-detector and to a difference amplifier which converts the light into a current; the computer is then able to determine the percent absorbance from this data and plot said absorbance vs wavelength1. The resolution of this reading (how accurately two different points can be measured) depends on the components themselves and will be explored in the following section.
Components of the Varian Cary 100 and their Functions
i) Source:
The source of light in a UV-spectrometer is typically a set of two lamps: a tungsten/halogen lamp for the visible wavelength, and a deuterium lamp for the UV region.
ii) Monochrometer:
1. The entrance slit determines what wavelengths are included; the wider the slit, the greater the threshold is for wavelength of light that can pass through.
2. The light then reflects off of a collimating mirror, which is specifically designed with a focal point such that when multiple rays of light reflect off of it, they are parallel after reflection.
3. The light then reflects off of an optical element known as grating, or diffraction grating. The grating is etched with corrugated slits of specific and constant measurements and the grating is typically coated with metal afterwards; when light of several different wavelengths comes into contact with it, each wavelength reflects at a different angle and is therefore separated and travels at a different trajectory. Grating can be rotated, and doing so alters the trajectory of the different wavelength.
4. The light then reflects off of the focusing mirror. The angle of reflection for each individual wavelength of light depends on its trajectory after reflection off of the grating; unlike the collimating mirror, they different wavelengths are not parallel. Multiple focusing mirrors are used in a spectrometer, and any unlabeled reflective surface in figure three should be assumed to be a focusing mirror.
5.. The light then travels to the exit slit. As each wavelength of light follows a different trajectory at this point, each wavelength arrives at a different point in the slit plane. Since the length between the focusing mirror and the exit slit is finite, it is impossible for a slit to allow only one specific wavelength of light through as there will always be some overlap; i.e., a beam of light with a wavelength of only .01 nm higher than the wavelength desired for analysis might have a similar enough trajectory so that, over the distance between the focusing mirror and the exit slit it fails to separate enough to avoid arriving at the slit opening.

Figure 2, outline of a monochromater
iii) Beam Splitter:
The purpose of a beam splitter in a double beam spectrometer is to direct the light to either the sample cell or the reference cell; the rotating chopper used in this model is a circle with alternating quadrants of either transparent or reflective surface. Separating each chopper from each cell is another focusing mirror to further guide the beams. As the name indicates, the coppers can rotate during analysis to direct the light to the desired sample.
iv) Detector:
The purpose of the detector is to convert the resulting beam of light into a current; this model uses a photodetector tube for this purpose. The photocathode of this device is covered with a photosensitive surface which releases an electron upon absorption of a photon which is amplified by a dynode chain. At the end of this chain is an anode. Over a large enough range, the current from anode to ground is proportional to the photoelectron flux generated by the photocathode. Software then calculates the absorbance from this data and it is presented in graph form.
III. Procedure.
Solutions analyzed
The solutions shown in table 1 (next page) were prepared. They were transferred to a cuvette of pathlength 1cm and then analyzed via UV-Vis spectroscopy from 650 nm to 200 nm, as outlined in the previous sections. From the data, lambda max was determined and the extinction coefficient was calculated via the handwritten method shown below:
IV. Results and Discussion
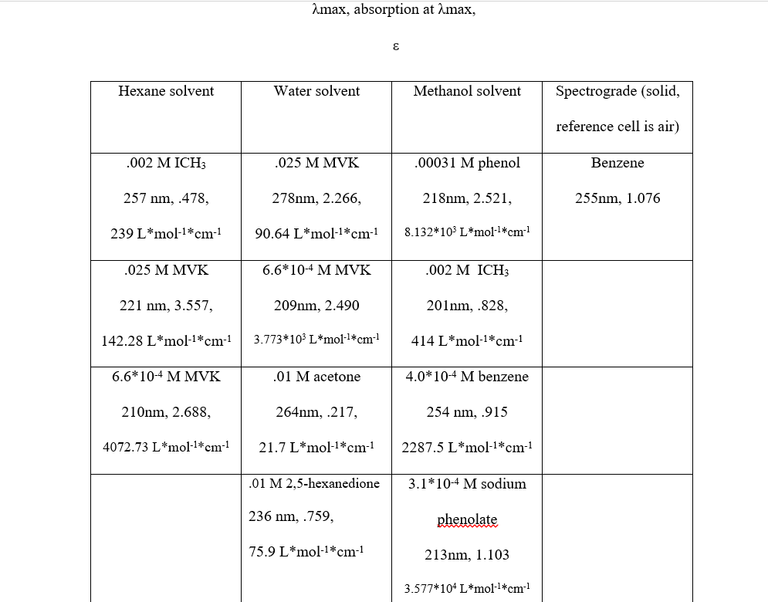
Table 2 shows the λmax values, absorption, concentration, solvent, and extinction coefficient of each sample. The λmax values, electron transitions, solvent and concentration effects are as follows:
- Benzene- The λmax values of 255 nm and 254 nm for the two benzene solutions are indicative of a π- π* transition, which is perfectly reasonable given that it is unsaturated and conjugated.
- MVK- From the data, a 69 nm blue shift occurs when diluting the MVK/water solution, and an 11 nm blue shift occurs when diluting MVK/hexane solution. The absorbance’s of both MVK/water solutions were less than the absorbance of each corresponding MVK/hexane solution (though relatively little in the case of the weaker solutions). The .025M solutions differed significantly in λmax values, while the .0006M solutions had very similar λmax values. From these facts, it was concluded that all of the MVK solutions experience both a concentration effect and a solvent effect and that the transitions for all of them were π- π* transitions. π- π* transitions, which can occur at wavelengths between 200-700 nm, frequently experience a redshift3 in polar solvents, which explains the 278 nm peak in the .025M MVK/water solution vs the 221 nm peak in the .025M MVK/hexane solution. The extinction coefficients for π- π* transitions in dilute solutions typically fall between 1000 and10000, well within the range of the .00066M solutions. Judging the extinction coefficients of the higher concentration MVK solutions leaves two options: either they actually experienced an n -π* transition or concentration effect and, in the case of the MVK/water solution, solvent effect greatly lowered the energy gap between the π -π* orbitals. An n -π* transition is very unlikely for two reasons; the first being that the values for λmax were far too low (typically greater than 300) and the second being that n -π* transitions experience a blueshift in polar solvents3 wheras the peaks observed experienced a redshift, which is again indicative of a π -π* transition. The decreased values of for the .025 M MVK are to be expected as increasing the concentration of a solution decreases the value of .
- ICH3- The ICH3 solutions experienced an n- σ* transition and a massive blueshift occurred when switching the solvent from methanol to hexane, likely due to the nonpolar ICH3 (as iodine and carbon have the same electronegativity value, 2.5) being much less miscible in the polar methanol than it would be in hexane and subsequently experiencing no energy lowering interaction.
- Phenol and sodium phenolate- Both compounds had very similar λmax values indicative of π- π* transitions when read against . Both were the same concentration, but drastically different values of and therefore absorbance were observed. This is still being evaluated, as not only should sodium phenolate have a higher absorbance, its λmax value should be approximately 30 nm larger. Assuming the correct peak was taken, a possible reason for the discrepancy is peak broadening due to increased solvent interaction
- Acetone and 2,5-hexanedione: Both ketones experienced an n-π* transition due to their relatively low value, however the diketone has two carbonyl functional groups to participate in n-π* transition, explaining the increase in absorbance. Additionally, the diketone can react interact with the solvent, intermolecularly, or intramolecularly to decrease ground state energy via dipole-dipole interactions. Acetone is able to do the first two, but not the third.
V. Post Lab Questions
1-2: Covered in paper
3: Their peak absorbance’s usually occurs below 200 nm
4-5: Covered in paper
6: 1nm, which is very small as π -π* transitions experience a redshift in polar solvents; either an incorrect peak was taken or an error in the reading occurred
7-8: Covered in paper
VI: Citations
Skoog; Holler; Nieman. Principles Of Instrumental Analysis; 5th ed.
Golden, T. D.; Conrad, H. A. Insturmental Analysis Laboratory Notebook.
UV-Vis Absorption Spectroscopy - Theory. UV-Vis Absorption Spectroscopy - Theory, http://teaching.shu.ac.uk/hwb/chemistry/tutorials/molspec/uvvisab1.htm (accessed Feb 7, 2016).
Molecular Spectroscopy. Beer-Lambert Law, https://hplc.chem.shu.edu/new/undergrad/molec_spectr/lambert.html (accessed Feb 7, 2016).
UV-VIS theory. UV-VIS theory, http://www.chem.ucla.edu/~bacher/uv-vis/uv_vis_tetracyclone.html.html (accessed Feb 7, 2016).
Effect of Slit Width on Signal-to-Noise Ratio in Absorption Spectroscopy. Simulation of Effect of Slit Width on Signal-to-Noise Ratio in Absorption Spectroscopy, https://terpconnect.umd.edu/~toh/models/absslitwidth.html (accessed Feb 7, 2016).