ANEMIA - Un disturbo tanto comune quanto misconosciuto
Parte 2
Cari Steemians,
Mi scuso per essere così in ritardo con la tabella di marcia, pubblicando solo ora la seconda parte di un articolo cominciato un mese fa ma.
Per chi si fosse perso l'articolo precedente e fosse interessato all'argomento, può leggere qui la prima parte della trattazione(sarei ben felice di ricevere vostri commenti/consigli/pareri e,se qualcosa dovesse essere eventualmente poco chiaro, anche domande!)
Ma ora non perdiamoci troppo in chiacchiere e partiamo subito parlando delle Anemie del III Gruppo!
III Gruppo - Ridotta sintesi emoglobinica
Le anemie del III Gruppo sono caratterizzate da riduzione della sintesi di emoglobina,per cui i globuli rossi risultano ipocromici(contenendo meno emoglobina risultano anche meno "coloriti") e microcitici(i precursori degli eritrociti vanno incontro a più divisioni mitotiche generando,infine,eritrociti dalle dimensioni ridotte).
E' un gruppo di anemie piuttosto importante, non perché gli altri siano meno importanti ma perché del III Gruppo fanno parte due delle più comuni anemie: l'Anemia sideropenica e la Talassemia(se,infatti, solo pochissimi di noi avevano precedentemente sentito parlare dell'anemia di Joseph-Blackfan-Diamond chi di noi non ha mai sentito parlare di anemia da carenza di ferro e talassemia?)
1.Anemia sideropenica(o da carenza marziale)
L'anemia sideropenica è la forma di anemia più frequente in assoluto.
Si manifesta quando nel nostro organismo c'è una carenza di ferro, essenziale per la sintesi del gruppo eme, che è in pratica la parte dell'emoglobina capace di legare l'ossigeno
(la parola emoglobina viene infatti da eme + globina, in cui l'eme, come abbiamo detto, è essenzialmente la parte legante l'ossigeno, mentre la globina è l'infrastruttura proteica all'interno della quale è collocato il gruppo eme. In particolare, nell'emoglobina abbiamo 4 strutture di questo tipo - eme + globina - legate tra loro, formando quello che viene chiamato un tetramero)
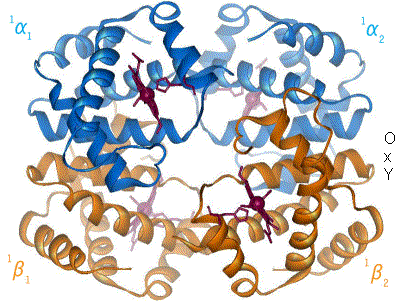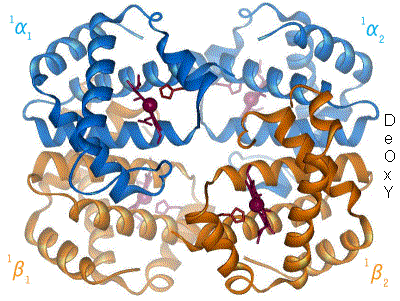
Immagine CC0 Creative Commons - Struttura dell'emoglobina adulta, che presenta 2 catene globiniche di tipo α (in blu) e due catene globiniche di tipo β (in arancione). I gruppi eme si trovano al centro delle catene globiniche e sono evidenziati in rosso. E',inoltre,mostrato il passaggio dell'emoglobina dallo stato ossigenato a quello deossigenato.
Una carenza di ferro può verificarsi per:
1. Ridotto apporto, per motivi dietetici o per cause organiche(per esempio in caso di acloridria gastrica, condizione in cui c'è un'insufficiente secrezione di HCl da parte dello stomaco con compromissione del processo di acidificazione gastrica, essenziale per l'assorbimento del ferro)
2. Aumentato consumo o perdita, come avviene durante l'accrescimento, in gravidanza o in conseguenza di perdite ematiche di una certa entità(sia fisiologiche, come nel caso delle mestruazioni, che patologiche, come può succedere successivamente a un trauma).
Attenzione però:
Carenza di ferro non significa anemia sideropenica;
ci possono essere infatti persone con una carenza di ferro ma non anemiche, questo perché il nostro organismo, per continuare a sintetizzare emoglobina, sottrae il ferro ad altri processi metabolici che ne hanno bisogno. Solo in uno stato di avanzata negativizzazione del bilancio del ferro avremo dunque anemia.
Una mancanza di ferro, quindi, oltre che con anemia si manifesta con altri segni e sintomi: coilonichia("unghia a cucchiaio"), cheilite angolare(lesioni agli angoli della bocca), glossite atrofica (infiammazione della lingua con degenerazione del suo rivestimento mucoso).
Immagine CC0 creative commons - Atomo di ferro
L'anemia sideropenica entra in diagnosi differenziale con le talassemie, poiché in entrambi i casi avremo un'anemia microcitica ed ipocromica; saremo tuttavia in grado di differenziare le due condizioni valutando, con i necessari esami di laboratorio, i depositi di ferro dell'organismo(ridotti nella prima, normali nella seconda) e grazie ad un parametro, l'RDW(red blood cell distribution width), che altro non è che la deviazione standard dell'MCV(Volume Corpuscolare Medio) e misura di quanto, in media, i volumi eritrocitari si discostano dalla media; questo parametro sarà elevato in caso di anemia sideropenica e ridotto in caso di talassemia.
La terapia dell'anemia sideropenica mira, innanzitutto, a risolvere le cause della carenza di ferro e poi a correggere la carenza stessa.
2. Talassemie
Le talassemie sono delle malattie congenite a carattere ereditario e conseguono ad un'alterazione quantitativa della sintesi di emoglobina(diversamente dalle emoglobinopatie che, come vedremo in seguito, sono caratterizzate da alterazioni qualitative).
A questo punto è necessario fare una breve digressione sulla struttura dell'emoglobina e sulle varie catene globiniche dalla cui combinazione derivano i diversi tipi di emoglobina per capire come dall'alterazione della sintesi di una catena globinica possano scaturire diversi effetti dannosi per il nostro organismo.
Come abbiamo detto prima, l'emoglobina è un tetramero, essendo formata da 4 catene globiniche legate tra loro.
Durante la nostra vita produciamo 6 tipi di catene globiniche, nominate con lettere dell'alfabeto greco: α,β,γ,δ,ε,ζ. Detto ciò, possiamo essenzialmente avere α e β talassemie, conseguenti, rispettivamente, a deficiente sintesi di catene α e di catene β, dovuta a mutazioni nei geni codificanti per queste catene. le catene β dell'emoglobina sono codificate da un singolo gene che si trova sul cromosoma 11( e quindi da 2 alleli presenti sui cromosomi omologhi). Sono state scoperte numerosissime mutazioni diverse che aboliscono o riducono la sintesi della catena globinica. 1. β talassemia maior (Morbo di Cooley): è la forma omozigote e più grave della β talassemia, nella quale abbiamo infatti abolizione completa della sintesi di catene β. La terapia è emotrasfusionale(che alla lunga può portare però delle complicanze ----> emosiderosi). 2. β talassemia intermedia: è in genere causata dalle "mutazioni talassemiche lievi", che permettono, cioè, una discreta sintesi di catene β. 3. β talassemia minor: forma eterozigote della β talassemia. Nella gran parte dei casi decorre in modo del tutto asintomatico. I pazienti, tutt'al più, presenteranno un "trait talassemico".
Le catene ε e ζ sono tipiche del periodo embrionale; la catena γ, insieme alla catena α, forma invece l'HbF(emoglobina fetale), la cui formula è α2γ2; la catena β comincia ad essere sintetizzata poco prima della nascita e soppianterà poi la catena γ, formando, insieme alla catena α, l'HbA1, la principale emoglobina dell'adulto, la cui formula è appunto α2β2; la catena δ viene,infine, sintetizzata nell'adulto in percentuali molto basse e forma l'HbA2, la cui formula è α2δ2.β talassemie:
Il fenotipo dei pazienti dipenderà, dunque, dalle condizioni di omozigosi, eterozigosi o eterozigosi composta che si vengono a creare e dal tipo di mutazione ereditata.
In base a ciò distinguiamo:
L'assenza di catene β porta ad un'impedita sintesi di HbA1 e, di conseguenza, ci sarà un eccesso di catene α libere, che continuano ad essere prodotte normalmente.
Le catene α in eccesso tendono, poi, a precipitare e ciò danneggia seriamente la membrana plasmatica degli eritrociti, che vanno incontro a lisi o comunque presentano una sopravvivenza molto ridotta.
Una condizione del genere, ovviamente, porterà ad una grave anemia, che si manifesta in pazienti pediatrici che presenteranno ittero, epatosplenomegalia ed alterazioni ossee( dovute all'iperplasia midollare indotta da un aumento dell'EPO, prodotta in risposta all'ipossia che si viene ad instaurare).
In questo caso la necessità di emotrasfusioni può essere nulla o scarsa.
Il motivo per il quale si deve però diagnosticare una β talassemia minor è eugenetico: infatti, dall'unione di due portatori sani( che non presentano né segni né sintomi di malattia) può nascere un bambino con β talassemia maior, e quindi gravemente malato.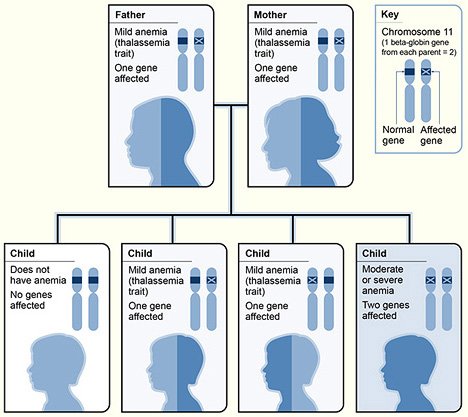
Immagine CC0 creative commons
α talassemie:
per quanto riguarda la catena α dell'emoglobina, abbiamo 2 geni,localizzati sul cromosoma 16, che codificano per questa catena globinica( e quindi 4 alleli sui cromosomi omologhi).
Sapendo questo, possiamo trovarci di fronte alle seguenti situazioni:
1. α talassemia 1: in questo caso abbiamo la deficienza di entrambi i geni per le catene α: se avviene su un solo cromosoma, siamo in eterozigosi, se avviene, invece, su entrambi i cromosomi siamo in omozigosi.
Allo stato eterozigote abbiamo un quadro clinico assimilabile a quello della β talassemia minor( le catene α sono prodotte a sufficienza per garantire al paziente una vita normale).
Allo stato omozigote, invece, l' α talassemia 1 è incompatibile con la vita,( le catene α sono assenti e non si può produrre nessun tipo di emoglobina fisiologica) e la morte avviene durante la gravidanza, a causa della formazione esclusivamente di un tipo patologico di emoglobina, l'Hb Bart(formata da un tetramero di catene γ, che normalmente, insieme alle catene α, formano l' HbF), che non rilascia adeguatamente l'ossigeno ai tessuti.
2. α talassemia 2: in questo caso abbiamo la deficienza di un solo gene per la sintesi della catena α e, come per l'α talassemia di tipo 1, abbiamo eterozigosi quando il difetto riguarda uno solo dei due cromosomi 16 ed omozigosi quando il difetto interessa entrambi i cromosomi.
I soggetti, però, non sono anemici in nessuna delle due condizioni, presentando, al massimo, globuli rossi ipocromici e microcitici.
3. Malattia da HbH( doppia eterozigosi α talassemia 1/α talassemia 2):
i pazienti affetti da questa forma di α talassemia hanno un solo gene per le catene α funzionante, il che porta ad un eccesso di catene β libere.
Le catene β in eccesso si aggregano e formano l'HbH(β4), un' Hb instabile che precipita e danneggia l'eritrocita.
L'anemia sarà severa o moderata.
Dovremmo ora procedere con il IV Gruppo di anemie, ma mi sono scoperto più prolisso di quel che credevo! Vi do quindi appuntamento al prossimo articolo, nel quale completeremo il nostro discorso sulle anemie!
Come sempre vi invito a pormi delle domande se qualcosa fosse poco chiaro e a darmi dei consigli se doveste averne: sono sempre ben accetti!
Buona lettura e al prossimo articolo!

Logo creato da @ilvacca

Immagine CC0 Creative Commons, si ringrazia @mrazura per il logo ITASTEM.
CLICK HERE AND VOTE FOR DAVINCI.WITNESS
Bibliografia:
- Tura S. Lezioni di Ematologia. Società editrice Esculapio,2003.
i have anemia. good blog
Thanks 😊
Kami telah upvote..