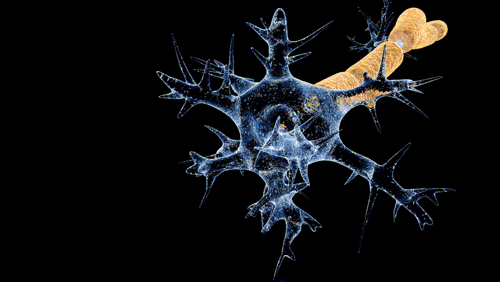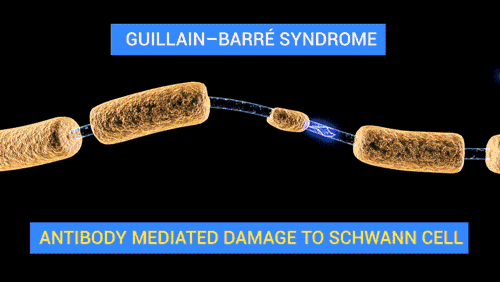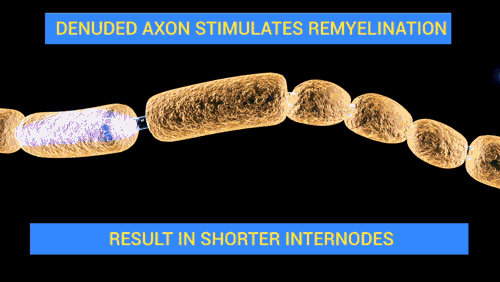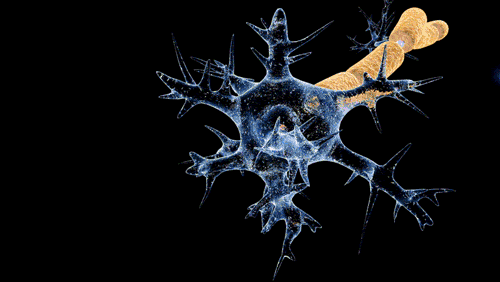
De Doctor Jana - http://docjana.com/#/gbs; http://www.patreon.com/posts/guillain-barre-4374004, CC BY 4.0, Enlace
Hola, amigos steemianos. En esta ocasión hablaremos sobre el Síndrome de Guillain-Barré (SGB), pero considerando que forma parte de las neuropatías, es pertinente hablar un poco de estas enfermedades.
Necesario conocer: El sistema nervioso tiene un componente central y uno periférico. El central lo constituyen el encéfalo, el tallo cerebral y la médula espinal que se encuentran protegidos por la bóveda craneal y los cuerpos vertebrales. Por otra parte, el periférico es aquel que se proyecta fuera del sistema nervioso central e inerva los miembros y órganos del cuerpo. A diferencia de los anteriores, no están protegidos por estructuras óseas. En este post, nos referiremos a los últimos mencionados.
En general, Neuropatía periférica se les llama a aquellos trastornos de los nervios periféricos, sea cual sea su causa. Estas pueden clasificarse según el número de nervios afectados y la parte del nervio afectado.
- Mononeuropatías: se ve afectado un tronco nervioso único.
- Polineuropatías: afectan múltiples troncos nerviosos y su instauración es gradual. Se caracterizan por ser distales, preferentemente, también simétricos y generalizados.
- Radiculopatías: afectan a las raíces nerviosas y las polirradiculopatías, al igual que la anterior, afectan las raíces nerviosas, pero de múltiples nervios, en este caso, de forma consecutiva.
- Plexopatías: afectan al plexo nervioso. Pueden ser traumáticas, compresivas o disinmunitarias.
La primera manifestación clínica de las neuropatías, en general, son los trastornos sensitivos. Los trastornos motores se caracterizan por tener signos de afectación de la segunda motoneurona y dependiendo del tipo de neuropatía, habrá atrofia muscular, sobre todo en las afectaciones axonales y poco frecuente en las enfermedades desmielinizantes. Los trastornos autonómicos incluyen hipotensión ortostática, estreñimiento, impotencia retención urinaria, diarrea, entre otros.
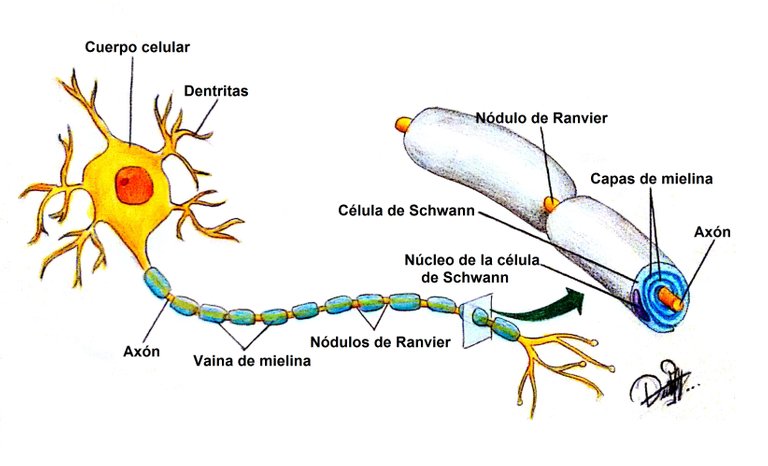
Dibujo de las partes de la neurona elaborado por el autor
RECUENTO HISTÓRICO

Georges Guillain (1876-1961) De Ouvrage : L'informateur des aliénistes et des neurologistes, Paris : Delarue, 1923, Dominio público, Enlace
Entrando en materia, el SGB es una polirradiculoneuropatía inflamatoria aguda de origen inmunológico. Se le dio su nombre en honor a George Guillain y Jean-Alexandre Barré, quienes la describieron por primera vez en 1916, durante la primera guerra mundial como una debilidad aguda y progresiva de las extremidades. El líquido cefalorraquídeo tenía características poco comunes con elevación de la albúmina, sin reacción linfocítica y recuperación total.
En los siguientes años, lo llamaron Síndrome de Landry-Guillain-Barré, ya que Landry describió en 1859 manifestaciones similares de esta enfermedad, pero mortales; parálisis ascendente en todos los miembros, debilidad de lis músculos faciales, laríngeos, mandibulares y del diafragma.
En el siglo pasado, Haymaker y Kemohan identificaron en 50 casos mortales que el edema del nervio destruía la vaina de mielina y que posteriormente, se producía el infiltrado linfocítico. Además, se produjeron muchos avances durante ese siglo que han sido de gran ayuda para seguir conociendo esta enfermedad; sus características, manifestaciones, pronóstico y tratamiento.
El SGB, generalmente, es una lesión precedida por un proceso infeccioso agudo respiratorio o gastrointestinal. Se ha asociado con múltiples agentes, entre los cuales destacan Campylobacter jejuni, alguno de los virus del herpes, citomegalovirus, virus de Epstein-Bar, Haemphylus influenzae, Mycoplasma pneumoniae, hepatitis (A, B y E) y más recientemente, Zika. También la aplicación de la vacuna contra la gripe porcina (influenza) en su momento se vinculó a esta enfermedad.
INMUNOPATOGENIA
La Polirradiculopatia desmielinizante inflamatoria aguda es la más común y la mejor estudiada de las variantes del SGB. El mecanismo fisiopatológico no está totalmente esclarecido, pero el origen autoinmunitario es la teoría más apoyada y esta se aplica a todos los subtipos.
Datos circunstanciales plantean que todos los casos de SGB son producto de respuestas inmunitarias contra antígenos extraños que se desvían al sistema nervioso por un mecanismo de semejanza molecular. Los objetivos nerviosos que se sugieren son los gangliósidos. Estos son glucoesfingolípidos complejos que poseen uno o más residuos de ácido siálico. Son indispensables para mantener la integridad de los axones, para la transmisión del impulso nervioso y en el proceso de mielinización. Suelen estar expuestos en la membrana plasmática de las células y esto los vuelve vulnerables a los ataques mediados por anticuerpos.
En el Hombre, abundan en el sistema nervioso y dentro de este, en sitios importantes como los nódulos de Ranvier. Se ha descrito la presencia de estos anticuerpos en un 20 a 50% de los casos de SGB, sobre todo contra GM1 en las variantes NAMA y NASMA (de las cuales se hablará más adelante) y en los casos precedidos de infección por C. jejuni (que además de estar asociada a estos anticuerpos, posee propiedades que induce la diferenciación de los linfocitos B y amplifica más aún la autoinmunidad humoral).
En más del 90% de los casos de Síndrome de Miller-Fischer se detectan anticuerpos anti-GQ1b IgG, los cuales no son detectados en otras formas de SGB (a menos que haya compromiso de los nervios motores extraoculares). Por otra parte, las concentraciones de IgG son mayores al inicio de la enfermedad.
En la polineuropatía desmielinizante inflamatoria aguda, el mecanismo fisiopatológico de la parálisis flácida y de las alteraciones sensitivas es el bloqueo de la conducción, es probable que mecanismos inmunitarios celulares y humorales contribuyan en la lesión de las células nerviosas. Los altos niveles de citocinas y receptores de citocinas en el suero y líquido cefalorraquídeo sugieren activación de los linfocitos T. Durante el daño a las células, se completa el depósito en la superficie externa de la célula de Schwann y la activación del complemento produce una desintegración de la vaina de mielina, además, atrae macrófagos que participan en la lesión a la mielina y los axones.
MANIFESTACIONES CLÍNICAS
Actualmente se conocen variantes de la enfermedad que se distinguen según sus características anatomopatológicas y electrodiagnósticas, principalmente.
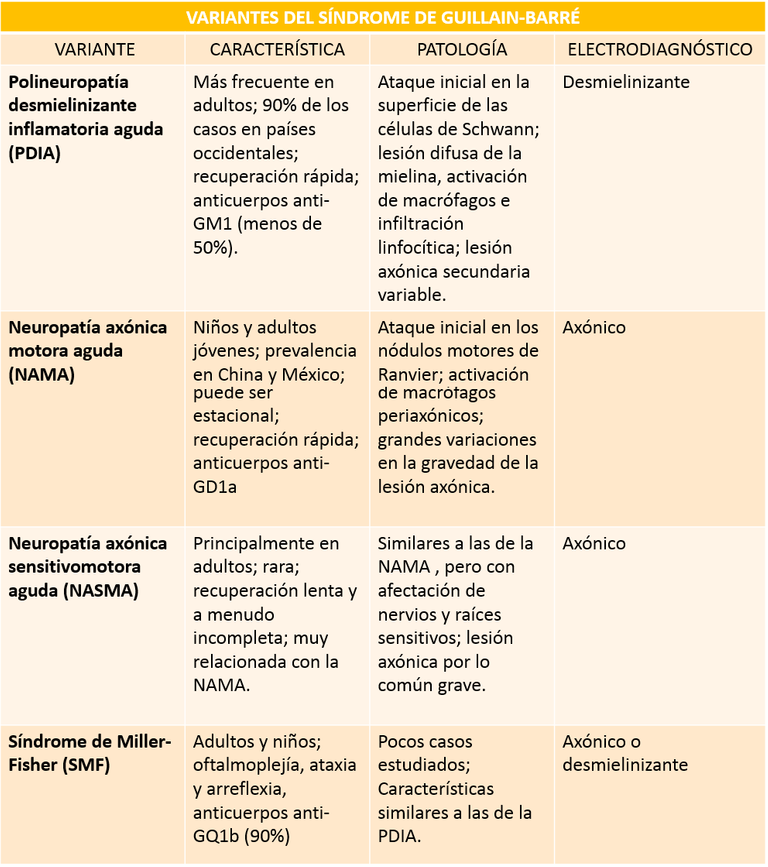
Cuadro explicativo elaborado por el autor
La progresión de los signos y síntomas del SGB va de 1 a 2 semanas. Los que se presentan con mayor frecuencia son:
- Dolor de espalda intenso (dorsalgia).
- Hormigueos (parestesias) en la porción distal de los miembros inferiores (en la mayoría de los casos) que progresa en sentido proximal.
De 1 a 2 días después de presentarse las parestesias, aparece:
- Debilidad muscular simétrica que limita actividades como levantarse de una silla o subir escaleras.
- Disminución o desaparición de los reflejos osteotendinosos profundos.
En el 50% de los casos, la manifestación inicial puede comprender la afectación de los músculos faciales y orofaríngeos produciendo dificultad respiratoria (disnea) que puede ser evidente a la inspección ya que el paciente presenta habla entrecortada y vacilante.
Cuando la parálisis progresa hasta afectar los músculos de la respiración, el paciente se complica y entra en insuficiencia respiratoria neuromuscular, haciendo necesaria la asistencia ventilatoria. Esto guarda relación con:
- Debilidad más grave al momento del ingreso.
- Progresión rápida.
- Presencia de debilidad facial o bulbar durante la primera semana de evolución.
La Disautonomía es otra complicación importante y puede ocurrir incluso en pacientes con SGB leve. Las manifestaciones habituales son:
- Fluctuaciones de la presión arterial
- Arritmias cardíacas
- Respuestas exageradas a fármacos.
- Hipotensión postural.
DIAGNÓSTICO
El síndrome de Guillain-Barré es una entidad descriptiva. Existen criterios (que se muestran al final de este segmento) que ayudan a realizar el diagnóstico o a descartarlo dependiendo de las manifestaciones clínicas que presente el paciente. Al identificar el patrón de parálisis de evolución rápida con arreflexia, ausencia de fiebre y otros síntomas sistémicos, además de los fenómenos antecedentes característicos, se establece el diagnóstico.
Los análisis complementarios son de suma importancia ya que aportan datos que apoyados en la clínica confirman el diagnóstico:
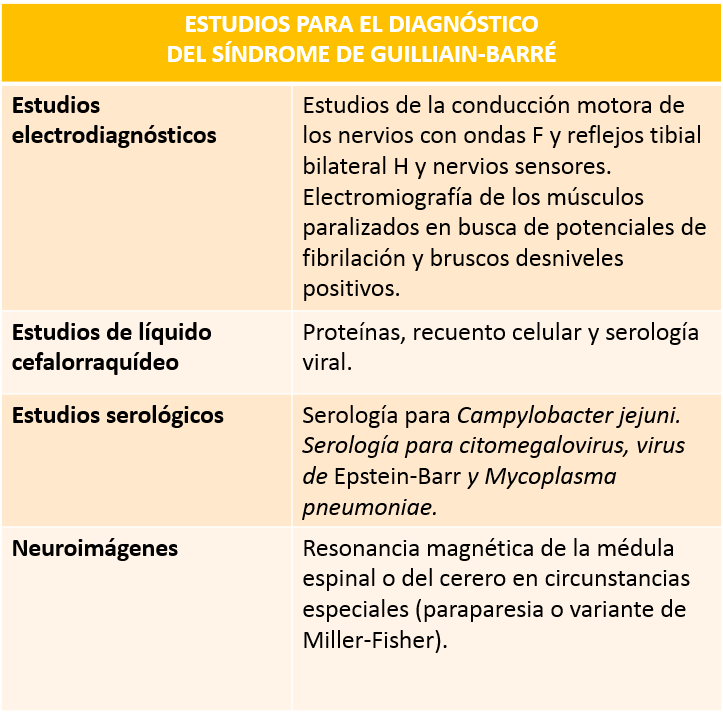
Cuadro explicativo realizado por el autor.
Los estudios electrofisiológicos son bastante útiles para el diagnóstico y ayudan a diferenciar (como se verá a continuación) entre las variantes axonal y desmielinizante del SGB.
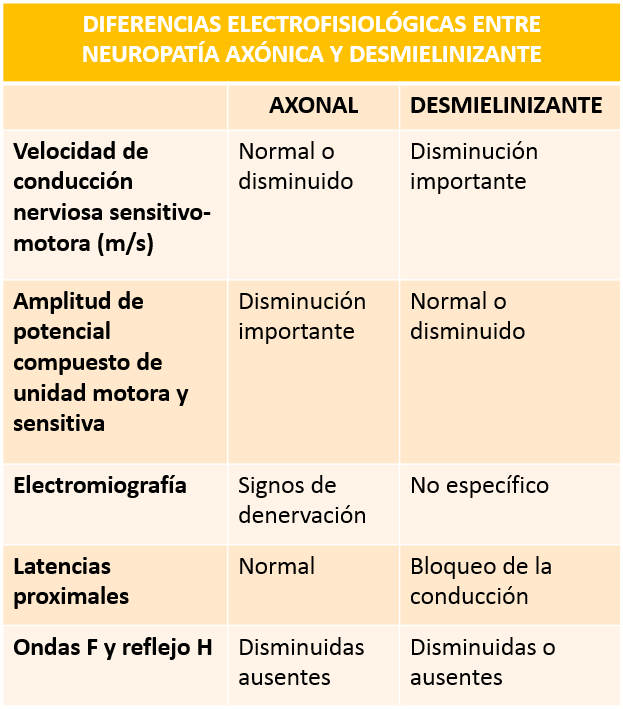
Cuadro diferencial elaborado por el autor.
La evaluación del líquido cefalorraquídeo posterior a las 48 horas de iniciados los síntomas, revelará una disociación albúminocitológica (altas concentraciones de proteínas con un recuento leucocitario normal) sin pleocitosis (presencia de células en el líquido cefalorraquídeo).
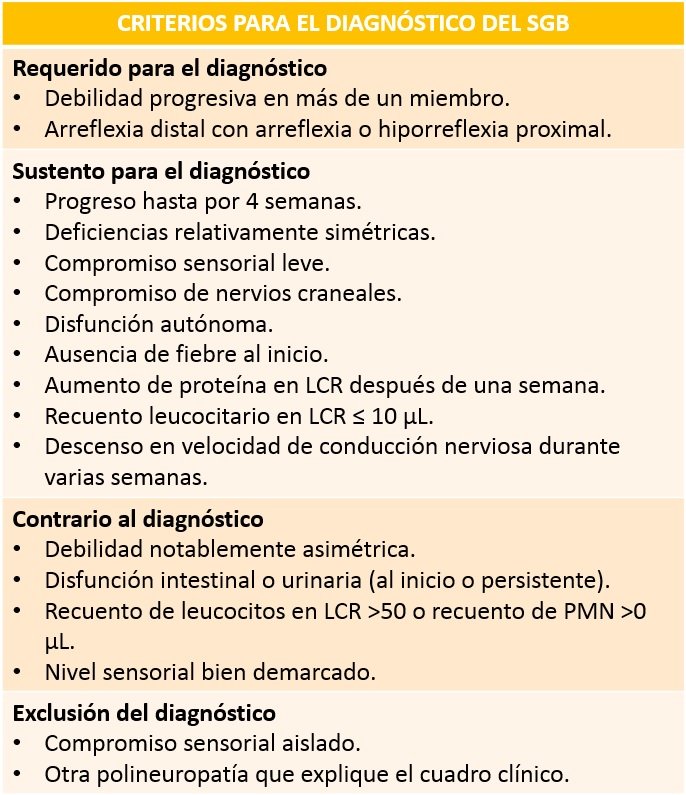
Tabla realizada por el autor
TRATAMIENTO
El tiempo es crucial para el inicio del tratamiento. Mientras más temprano sea el inicio de la terapia, mejor será el pronóstico de la enfermedad.
Inmunoterapia

Cuadro realizado por el autor
La plasmaféresis mejora la función supresora de las células T al eliminar los anticuerpos y otros componentes inflamatorios como el complemento.
Se ha demostrado que la IGIV y la plasmaféresis poseen una eficacia similar, aunque la primera produce menos complicaciones.
Tratamiento en la UCI
Uno de cada tres pacientes presentará un cuadro grave que amerite monitoreo estrecho e intubación endotraqueal. La evolución del SGB es bastante impredecible, lo cual dificulta su tratamiento.

Tabla realizada por el autor
Cuando la fuerza muscular diafragmática se recupere y se alcancen valores normales de las pruebas funcionales respiratorias, se deme empezar a retirar la ventilación mecánica. Éste debe realizarse cuanto antes ya que existen una gran cantidad de complicaciones relacionadas a la intubación prolongada.
El Síndrome de Guillain-barré es una neuropatía que puede llegar a ser mortal si no es diagnosticada y tratada a tiempo. Actualmente, se tienen conocimientos suficientes y se han desarrollado protocolos terapéuticos bastante eficaces y que han permitido disminuir las tasas de mortalidad por esta enfermedad.
REFERENCIAS
- Fauci AS, et al. Harrison’s principles of internal medicine. Vol 2. 19th ed. New York: McGraw Hill; 2015.
- Eelco F.M, et al. Síndrome de Guillain-Barré. Mayo Clin Proc. 2017; 92(3):467479.
- Allan H. et al- Principios de neurología de Adams y Victor. 8va edición. México, D.F.: McGraw Hill; 2007.
- Asbury AK, combalth DR, Assessment of current diagnostic criteria for Guillain-Barré syndrome. Ann Neurol. 10;27 (suppl.):S21-S24.

Muy interesante información @davt014, mi suegro sufrió esta enfermedad la supero ... pero fue muy fuerte lidiar con esto, es ocurrió hace como 5 años , hace dos años falleció pero con otro diagnostico. Gracias por publicar sobre esta enfermedad. Solo un resteem, que bueno que pude leer a tiempo para aunque sea resteemear, no se si esto se escribe así.
Gracias, lis. Aprecio bastante que te hayas tomado el tiempo de leer. Siento mucho lo de tu suegro.